Queste patologie sono descritte come una condizione patologica determinata da un precoce processo sinostosico delle suture craniche e questo fa sì che il cranio in alcuni settori diventi non espansibile, rigido, differentemente dal cervello che invece continua a crescere portando il soggetto ad uno stato di sofferenza come nei casi di ipertensione endocranica o sofferenza cerebrale. In questo caso il paziente deve essere sottoposto ad intervento chirurgico di decompressione encefalica, in maniera tempestiva. Le suture della volta cranica vengono considerate “zone di crescita ” che rispondono alla spinta dei tessuti intracranici (encefalo) ed intraorbitari (globi oculari). Normalmente le suture si chiudono in epoca post-natale (in un periodo che varia dai 3 mesi ai 40 anni) invece nelle craniostenosi la saldatura delle suture avviene nel periodo pre-natale. La ridotta o asimmetrica crescita craniofacciale che ne deriva determina alterazioni della forma delle strutture della volta e della base cranica. Questi quadri, secondo la gravità, possono determinare importanti anomalie funzionali a livello mentale, respiratorio, visivo, masticatorio, foniatrico, in particolare nelle forme che coinvolgono più suture. I quadri che interessano una sola sutura presentano solitamente una condizione clinica meno grave. Essendo la crescita cranica prevalente nel I anno di vita extrauterina, la patologia si presenta nei primi mesi di vita. La maggior parte delle craniostenosi coinvolge una sola sutura senza presentare anomalie in un'altra sede dell'organismo (craniostenosi semplici non sindromiche), altre, invece, possono interssare più suture senza anomalie a distanza (craniostenosi multiple non sindromiche).
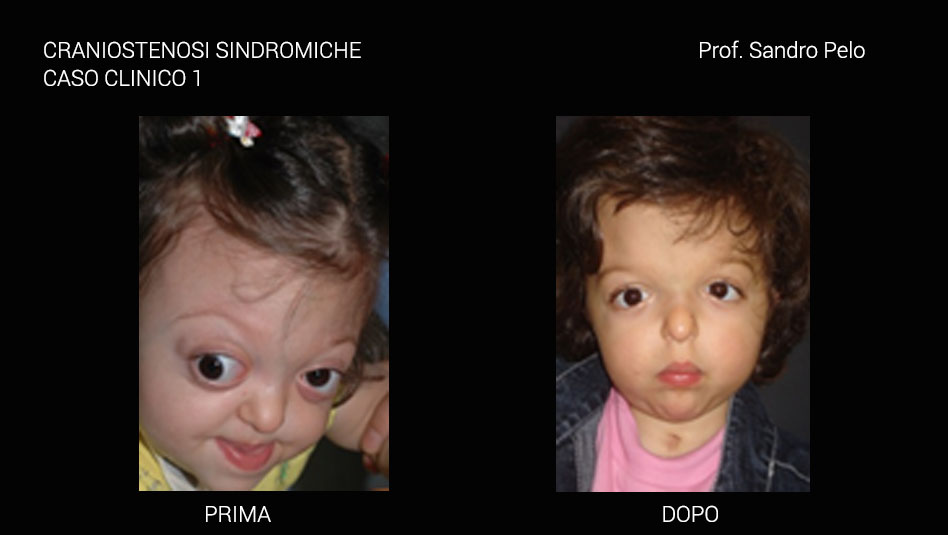
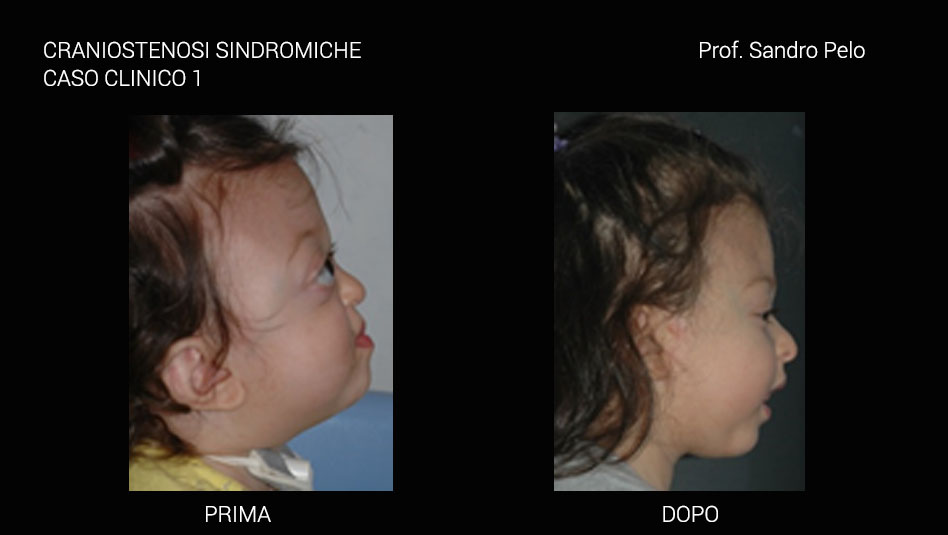
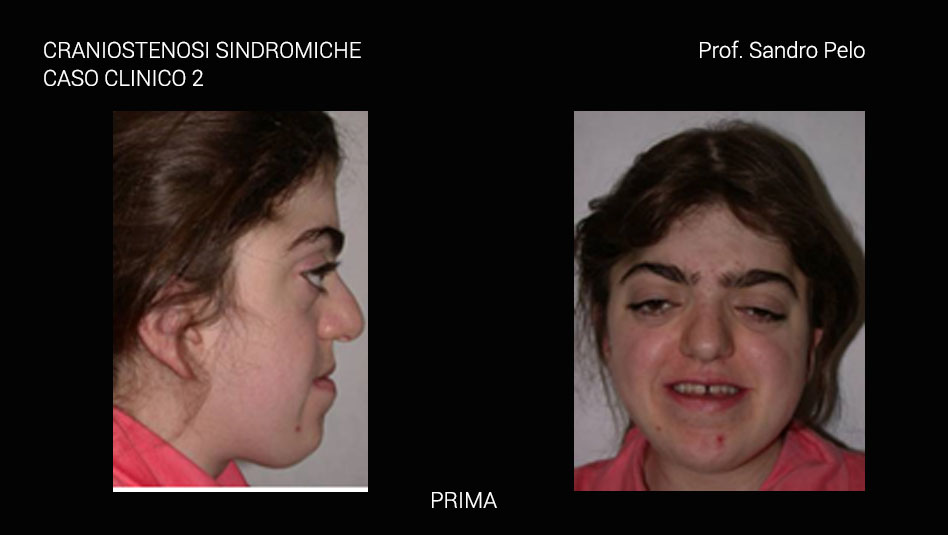
Una parte consistente di craniostenosi presenta, invece, il coinvolgimento di altri distretti dell'organismo determinando anomalie più o meno marcate (craniostenosi sindromiche per es. la sindrome di Crouzon e quella di Apert) ed in questo caso sono sempre multiple coinvolgendo più suture craniche. Le craniostenosi sindromiche sono caratterizzate da un arretramento della struttura fronto-naso-maxillo-orbitaria causando 4 problematiche fondamentali: a) sofferenza cerebrale b) problemi occlusali ( rapporto di III classe), masticatori, della deglutizione e della fonazione c) problemi respiratori (per riduzione degli spazi nasofarigei), con sonnolenza diurna, affaticamento, problemi comportamentali, incubi, ipossia ,difficoltà di concentrazione, iposviluppo mentale, iposviluppo somatico, ipertensione sistemica, ipertensione polmonare, morte nel sonno; d) exorbitismo o esoftalmo, per riduzione del volume orbitario, che può determinare lesioni corneali secondarie all' incompetenza palpebrale che ne deriva, cheratiti, papilledema, danni al nervo ottico fino ad arrivare a veri e propri traumi del globo oculare con lussazione dello stesso. Infine, talune forme di craniosinostosi possono presentarsi con ipertelorismo o distopia condizioni entrambe che si traducono oltre che in una importante alterazione della morfologia del viso anche in alterazioni della funzione visiva.
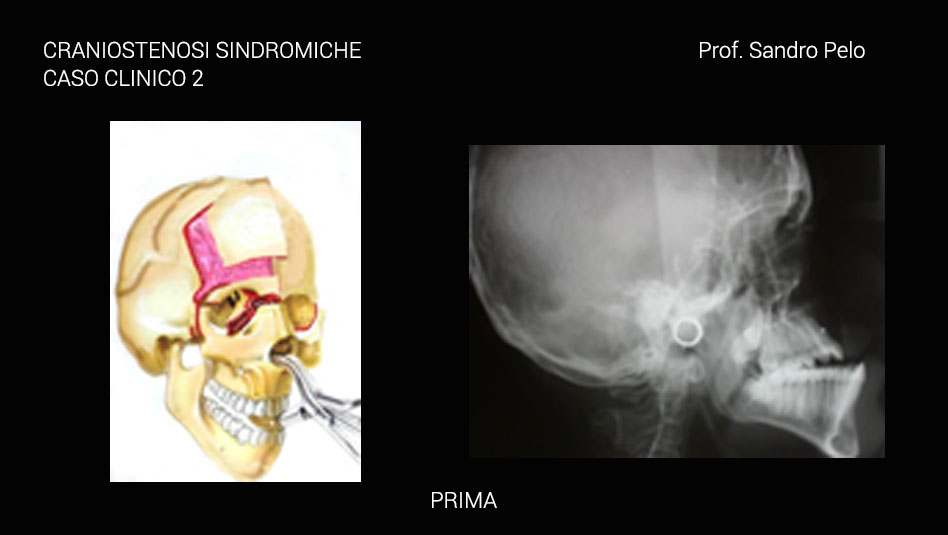
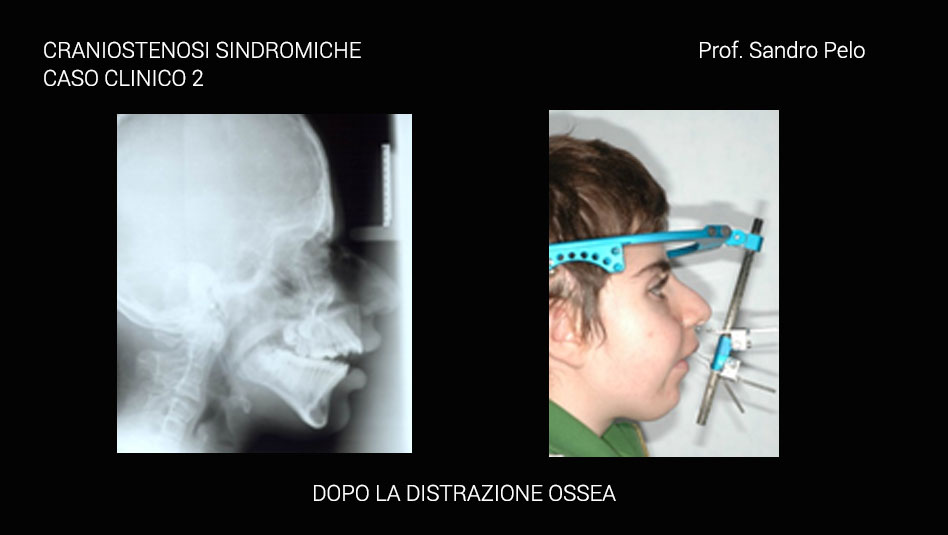
Dalla descrizione di tale patologia, quindi, è facile intuire come il timing terapeutico sarà differente a seconda della gravità della situazione clinica in rapporto da una parte con il rischio sulle funzioni e dall’altra con il timing di crescita dei diversi segmenti del complesso cranio facciale; in altri termini si tende a ritardare l’intervento correttivo fino a quando il segmento scheletrico coinvolto dalla patologia malformativa non è giunto a termine di crescita per evitare l’insorgere di recidive e la necessità di reinterventi; ovviamente se è presente il rischio di compromettere per sempre una funzione si interviene immediatamente. A tal fine è utile ricordare come la teca cranica raggiunge il 95% del suo sviluppo complessivo intorno ai 5 anni, la base cranica intorno ai 7 anni, il mascellare superiore verso i 15 anni e la mandibola solo intorno ai 18 anni cioè alla fine del processo di crescita complessivo dell’individuo. Anche l’interazione psicologica tra il bambino portatore della malformazione e gli altri bambini può condizionare i tempi di trattamento quando la situazione di “ mostruosità” in cui versa il piccolo paziente può dar vita a fenomeni di ghettizzazione che potrebbero condizionare la psiche del bambino per il resto della vita.
La prevalenza delle craniostenosi si aggira intorno ad 1 caso ogni 2000 – 3000 nascite. Sulla base delle suture coinvolte e dei quadri clinici relativi, ne sono state identificate e classificate diverse tipologie.
La causa primaria di tale patologia è stata riscontrata in una alterazione della trasmissione degli stimoli (stimoli espansivi legati alla crescita volumetrica del cervello) che la dura madre dovrebbe esercitare a livello delle suture craniche; studi genetici hanno permesso di stabilire un'importante correlazione tra anomalie geniche e diverse craniosinostosi, in particolare sindromiche: le mutazioni maggiormente note risultano a carico dei geni FGFR (1,2 e 3), MSX2 e TWIST. In particolare 7 importanti quadri sindromici sono stati associati ad anomalie del gene codificante per il recettore del FGF dimostrando come tale fattore di crescita risulti cruciale nello sviluppo delle ossa craniche.
Craniostenosi semplici
1) posizione normale del vomere e del temporale senza deviazione della piramide nasale;
2) posizione normale del vomere e dislocazione anteriore grave del temporale;
3) lieve deviazione del vomere e grave dislocamento anteriore del temporale;
4) deviazione del vomere, dislocamento anteriore del temporale ed appiattimento dell’occipitale.
Craniostenosi sindromiche
- Sindrome di Crouzon
- Sindrome di Apert o Acrocefalosindattilia
- Sindrome di Pfeiffer
- Sindrome di Saette-Chotzen
L’incidenza della sindrome Crouzon è stimata in 1 bambino su 25.000 nati vivi e rappresenta il 4.5% delle craniostenosi. È una patologia genetica trasmessa in maniera autosomica dominante ed è solo sporadica per mutazioni de novo.
Il gene responsabile è stato comprovato essere il FGFR2 (recettore del fattore di crescita del fibroblasto 2).
Allo stato attuale delle conoscenze, la diagnosi prenatale può essere ottenuta solo dal III trimestre di gravidanza mediante l’ecografia.
La principale malformazione consiste nella prematura e progressiva craniosinostosi, spesso presente alla nascita e che si completa verso i due o tre anni. Il tipo di deformità cranica è variabile, a causa di una imprevedibile sequenza di ordine, severità e grado di progressione della sinostosi suturale. La combinazione tra i vari tipi di chiusura delle suture può portare ad una serie difficilmente classificabile di malformazioni del capo. Nonostante ciò che il 96 - 98% dei pazienti presenta prematura chiusura delle suture coronale e sagittale, mentre il 79 % di essi mostra, anche, il coinvolgimento della sutura lambdoidea. Pertanto la forma del cranio risulterà con la volta parietale accorciata, fronte scoscesa e regione occipitale appiattita.
La precoce chiusura delle suture dà luogo a segni e sintomi neurocranici, presenti in buona parte dei casi, dovuti ad una severa limitazione all’espansione dell’encefalo.
L’ipoplasia del terzo medio della faccia risulta da una notevole varietà di fattori, che interferiscono con la crescita della base e della volta cranica. Aberrazioni dello sviluppo, chiusura prematura delle suture, aumento della pressione intracranica o combinazione di questi fattori possono portare ad una ipoplasia dell’osso frontale, dello sfenoide, dell’etmoide e del mascellare. La risultante retrusione del terzo medio della faccia e l’insufficiente profondità delle orbite sono segni caratteristici della sindrome di Crouzon.
La morfologia facciale è caratterizzata da appiattimento del dorso nasale, dalla ipoplasia della cresta sopraorbitaria, dall’ipertelorismo con appiattimento delle orbite e conseguente proptosi, naso a “becco di pappagallo” e zigomi ipoplasici. L’ipertelorismo e l’esoftalmo risultano sempre presenti in questa sindrome, così come lo strabismo, che può essere esotropico (deviato in dentro) o ipertrofico (deviato in alto). Inoltre, lo strabismo può essere deviato in basso o avere una caratteristica forma a V.
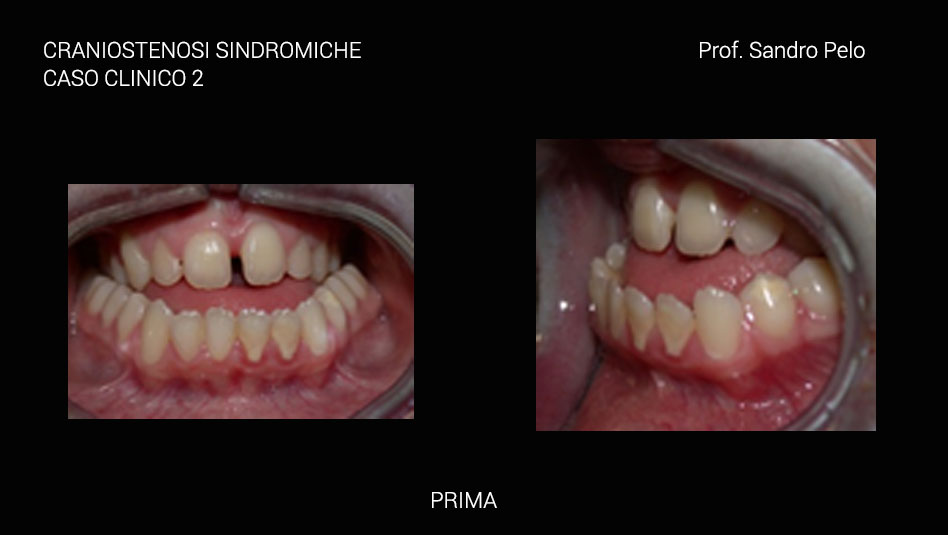
La carie è frequente. Il palato è stretto e inarcato, si possono avere alterazioni dell’eruzione dei denti e malocclusione scheletrica di III classe che accentua ancora di più la malformazione facciale. Atresia coanale e stenosi del canale uditivo associata a malformazioni della posizione dell’orecchio esterno e del canale uditivo, sono abbastanza frequenti. Altre caratteristiche fenotipiche della malformazione sono ossa del femore e dell’omero brevi ed in qualche raro caso è descritta la sindattilia parziale. Sporadicamente viene segnalata sclera blu, cataratta, ectopia del cristallino, glaucoma, coloboma dell’iride, megalocornea, microcornea, nistagmo e ipoplasia del nervo ottico.
La diagnosi può essere prenatale dal III trimestre di gravidanza mediante ecografia.
La craniosinostosi è sempre bicoronale e nella maggior parte dei casi sporadica ma a volte autosomica dominante, condiziona l’ipoplasia del terzo medio associata a sindattilia degli arti superiori ed inferiori con tendenza alla fusione della componente ossea a carico del secondo, terzo e quarto dito.
A livello facciale si manifesta con distopia orbitaria con severo exorbitismo, rima palpebrale con flessura antimongoloide, retrusione della radice nasale, incompetenza labiale per insufficienza del labbro superiore, palato ogivale con schisi incompleta, sindrome progenica con open bite, ipoplasia del III medio facciale. Inoltre possono essere presenti ritardo mentale e psicomotorio, anomalie cardiovascolari e genitourinarie, alterazioni a livello della trachea. Una caratteristica frequente è l’acne cutanea intrattabile.
La trasmissione è autosomica dominante, con penetranza completa ed espressione variabile. E’ eterogenea: sono state descritte due localizzazioni, sul cromosoma 8p11 e 10q26. Sono stati riportati una singola mutazione ricorrente del recettore 1 del fattore di crescita dei fibroblasti (FGFR1) e molte altre mutazioni coinvolgenti il recettore 2 (FGFR2).
Durante il periodo prenatale, attraverso l’ecografia morfologica è possibile far diagnosi o avere il sospetto di sindrome di Pfeiffer. Il sospetto clinico può poi essere confermato dall’analisi molecolare: attraverso il sequenziamento del DNA è possibile identificare mutazioni specifiche della sindrome.
Si può presentare in 3 forme:
Sindrome classica di tipo 1: i bambini hanno ipoplasia della parte media del viso e anomalie delle mani e piedi. L’intelligenza è normale.
Sindrome di tipo 2 : è presente un cranio a forma di trifoglio, protposi, anomalie delle dita delle mani e dei piedi, anchilosi dell’articolazione del gomito , complicanze neurologiche e ritardo nello sviluppo. E’ la forma più grave.
Sindrome di tipo 3: non c’è il cranio a forma di trifoglio ma può essere presente una sovrapposizione di tutti e tre i sottotipi. I segni clinici comuni sono: zigomi piatti, ptosi, pollice bifido, fronte alta, ipertelorismo, radice del naso larga, mani corte, sinidattilia delle mani e dei piedi, simfalangia delle dita, palato stretto, collo corto.